DL for Crystalline Solids
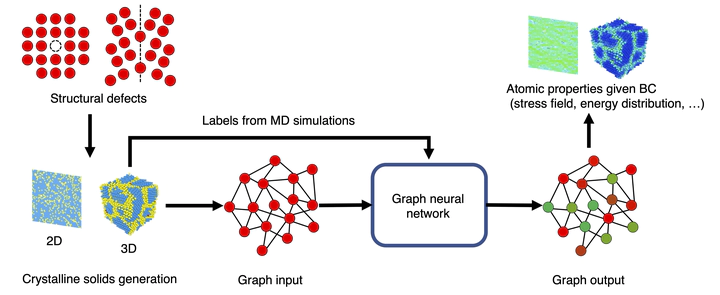
Structural defects are abundant in solids, and vital to the macroscopic materials properties. However, a defect-property linkage typically requires significant efforts from experiments or simulations, and often contains limited information due to the breadth of nanoscopic design space. Here we report a graph neural network (GNN)-based approach to achieve direct translation between mesoscale crystalline structures and atom-level properties, emphasizing the effects of structural defects. Our end-to-end method offers great performance and generality in predicting both atomic stress and potential energy of multiple systems with different defects. Furthermore, the approach also precisely captures derivative properties which strictly observe physical laws and reproduces evolution of properties with varying boundary conditions. By incorporating a genetic algorithm, we then design de novo atomic structures with optimum global properties and target local patterns. The method would significantly enhance the efficiency of evaluating atomic behaviors given structural imperfections and accelerates the design process at the meso-level.
Zhenze Yang
PhD student
My research interests include computational materials science, multiscale modeling and machine learning