Screening and Understanding Li Adsorption on Two-Dimensional Metallic Materials by Learning Physics and Physics-Simplified Learning
Abstract
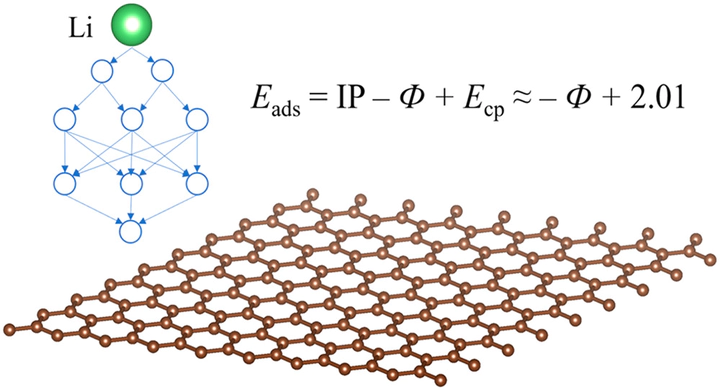
Understanding and broad screening Li interaction energetics with surfaces are key to the development of materials for a wide range of applications including Li-based electrochemical capacitors, Li sensors, Li separation membranes, and Li-ion batteries. In this work, we build a high-throughput screening scheme to screen Li adsorption energetics on 2D metallic materials. First, density functional theory and graph convolution networks are utilized to calculate the minimum Li adsorption energies for some 2D metallic materials. The data is then used to find a dependence of the minimum Li adsorption energies on the sum of ionization potential, work function of the 2D metal, and coupling energy between Li+ and substrate, and the dependence is used to screen all 2D metallic materials. Physics-simplified learning by splitting the property into different contributions and learning or calculating each component is shown to have higher accuracy and transferability for machine learning of complex materials properties.
Zhenze Yang
PhD student
My research interests include computational materials science, multiscale modeling and machine learning